Importing data¶
Experimental interface
The visual interface for imports with uploads was added in the recent G-nom rework. It is functional but expected to undergo visual rennovations in the near future.
All imported data references a species NCBI ID, as outlined in the data hierarchy. The first step of every import is selecting the NCBI taxonomy ID in the dialogue seen below.
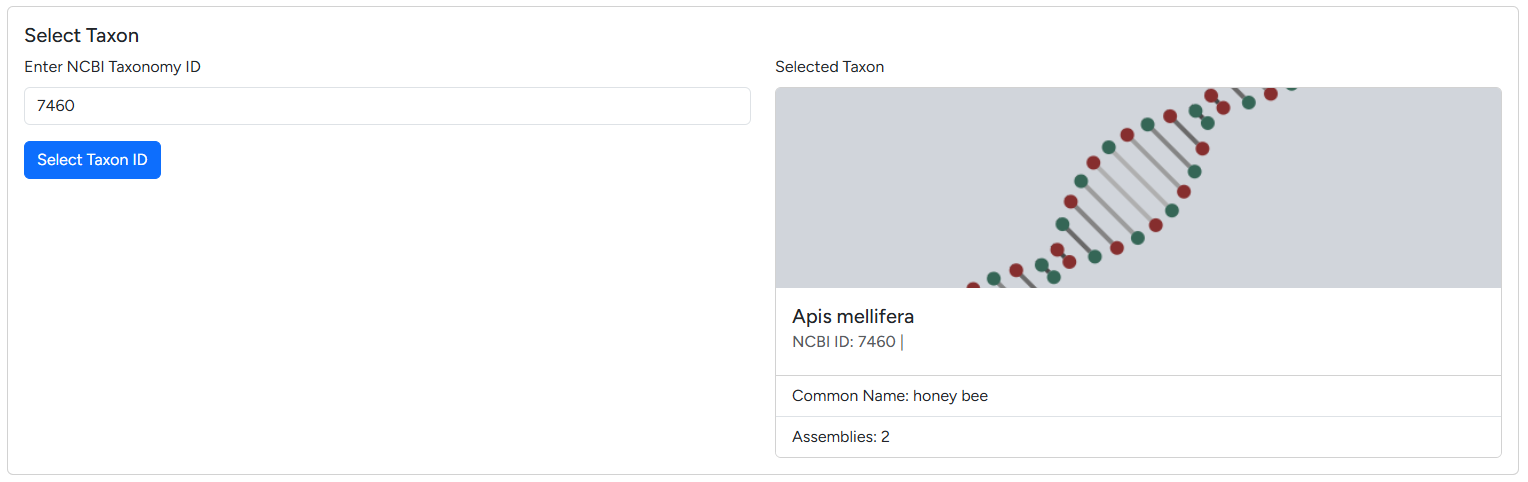
Taxonomy ID dialogue
Following the data hierarchy, your imported data is either a new assembly or references an existing assembly (e.g. mappings or annotations). The next dialogue allows you to decide between importing a new assembly or adding data for an exisiting one.
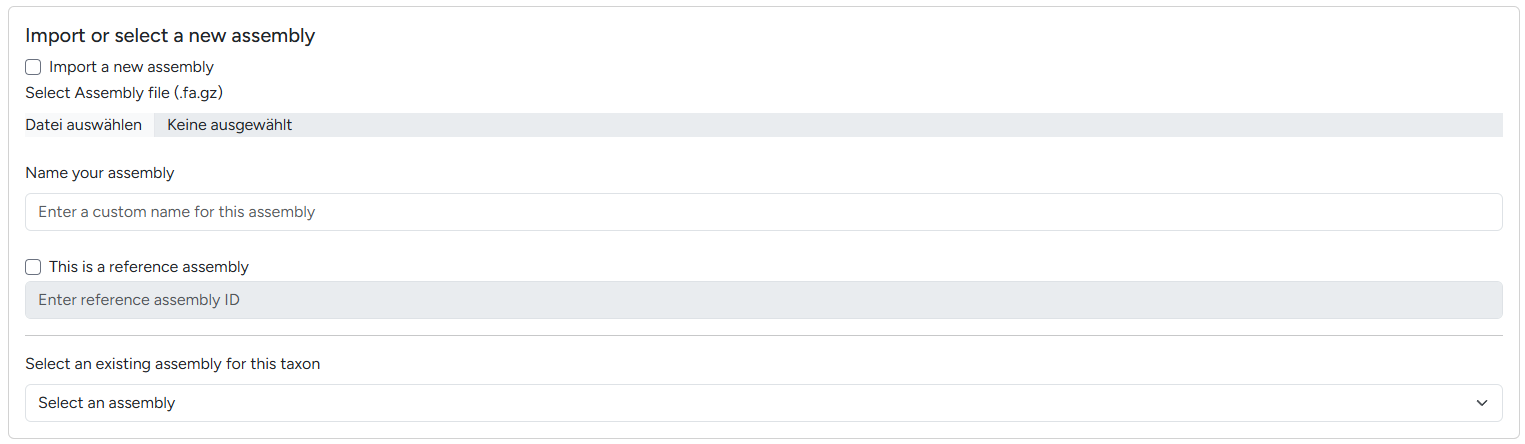
Switching between new / existing genome assembly
The following sections outline the import options available:
Importing Assemblies¶
Info
Future version of G-nom will automatically allow you to automatically import reference assemblies from IDs. You can track progress here.
flowchart TD
id1(Upload assembly) --> id2(Calculate assembly statistics)
id1(Upload assembly) --> id3(bgzip file for storage)
id3(bgzip file for storage) --> id4(Generate FASTA index)
id4(Generate FASTA index) --> id5(JBrowse import)
id5(JBrowse import) --> id6(Mark assembly completed)
id2(Calculate assembly statistics) --> id6(Mark assembly completed)- Specify the NCBI taxonomy ID of the species corresponding to your assembly as outline above
- Under Import or select a new assembly, tick the Import a new assembly tickbox
- Enter a name for your assembly, this will later be displayed in the assembly page header and on the assembly card
- If your assembly is a reference assembly sourced from a database like NCBI or GENCODE, specify the stable ID of the assembly in the reference assembly ID field
- At the bottom of the page click the start import button
Importing Annotations¶
flowchart TD
id1(Upload assembly) --> id2(gff3sort)
id2(gff3sort) --> id3(bgzip file for storage)
id3(bgzip file for storage) --> id4(Generate tabix adapters)
id4(generate tabix adapters) --> id5(Mark annotation complete)Importing Mappings¶
Warning
Mapping to larger genome can lead to large mapping files exceeding 20GB. Depending on your webserver configuration, you may encounter problems uploading. In this case, dispatch the import job manually in the server filesystem.
flowchart TD
id1(Upload mapping) --> id2(Compress if uncompressed)
id2(Compress if uncompressed) --> id3(Index)
id3(bgzip file for storage) --> id5(Mark mapping complete)- Specify the NCBI taxonomy ID of the species corresponding to your mapping
- Select a pre-existing assembly
- Under Import Analyses > Mapping, select a SAM or BAM file to upload and specify a custom name for the mapping. This name will be used as track label in the genome browser.
Importing BUSCO analyses¶
Info
The BUSCO analysis can be generated automatically using the G-nom core pipeline.
- Specify the NCBI taxonomy ID of the species corresponding to your mapping
- Select a pre-existing assembly
- In your BUSCO output locate the file matching
short_summary.specific.<lineage_dataset>.<output_folder>.json - Under Import Analyses > BUSCO, select the aforementioned file and specify a custom name for the analysis.
Importing fCAT analyses¶
Currently under development.
Importing Repeatmasker analyses¶
Currently under development.
Importing taXaminer analyses¶
Currently under development.